Home – Agile Molecule | Innovative Software & Agile Solutions
In a world where technology evolves at lightning speed, businesses need more than just software—they need agile partners who can anticipate change, adapt quickly, and deliver value with precision. Agile Molecule was founded with this belief at its core: to provide innovative software and agile solutions that empower organizations to thrive in dynamic environments.
Who We Are
Agile Molecule is a team of thinkers, builders, and problem-solvers who share a passion for technology and innovation. We specialize in delivering custom software and agile strategies that align with the unique needs of our clients. By combining technical expertise with adaptive processes, we ensure that every solution we create is both cutting-edge and practical.
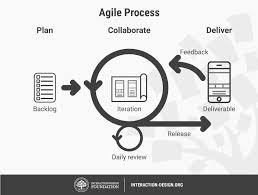
Our approach is rooted in the idea that agility is not just a methodology—it’s a mindset. It allows us to respond to shifting markets, evolving customer demands, and emerging technologies with confidence and creativity.
What We Offer
At Agile Molecule, we provide a comprehensive range of services designed to support organizations at every stage of their digital journey:
-
Custom Software Development – Tailored applications that address specific business challenges and scale with growth.
-
Agile Consulting – Guidance to help teams adopt and refine agile practices, improving collaboration and efficiency.
-
Technology Integration – Solutions that connect systems, streamline processes, and enhance performance across platforms.
-
Continuous Delivery & Support – Long-term partnerships that ensure solutions remain reliable, updated, and future-ready.
Each service is built on collaboration, ensuring that clients are involved in every step of the process.
Our Philosophy
What sets Agile Molecule apart is our philosophy of combining innovation with adaptability. We don’t just build software—we create solutions designed to evolve alongside our clients’ businesses. This means shorter development cycles, transparent communication, and iterative improvements that deliver value faster.
We believe that the best outcomes are achieved when technology, people, and processes work in harmony. That’s why we emphasize teamwork, user-centered design, and flexibility in every project.
Why Choose Agile Molecule?
Choosing Agile Molecule means choosing a partner that:
-
Listens first to understand your goals and challenges.
-
Collaborates closely to co-create solutions, not impose them.
-
Delivers quickly through agile methods, without compromising quality.
-
Plans for the future, ensuring scalability and resilience in every solution.
Our clients trust us because we don’t just deliver technology—we deliver confidence, efficiency, and competitive advantage.
Looking Ahead
The digital landscape will continue to change, but with Agile Molecule by your side, your organization will be ready to embrace opportunities and overcome challenges. Our mission is to help businesses innovate with confidence, adapt with agility, and achieve lasting success.
This site, Agile Molecule | Innovative Software & Agile Solutions, is your starting point to explore how we can work together. From custom development to agile transformation, we invite you to join us on a journey where technology becomes an enabler of growth and progress.
At Agile Molecule, we don’t just build software—we build the future.